FeatureCloud – Privacy preserving federated machine learning and block chaining for reduced cyber risks in a world of distributed healthcare
The digital revolution, in particular big data and artificial intelligence (AI) offer new opportunities to transform healthcare. However, it also harbors risks to the safety of sensitive clinical data stored in critical healthcare ICT infrastructure. In particular, data exchange over the internet is perceived insurmountable, posing a roadblock hampering big data based medical innovations.
With FeatureCloud, we chair a pan-European transformative AI development project, which implements a software toolkit for substantially reducing cyber risks to healthcare infrastructure by employing the world-wide first privacy-by-architecture approach, which has two key characteristics: (1) no sensitive data is sent through any communication channels, and (2) data is not stored in one central point of attack.
Web: https://featurecloud.eu
TiCoNE - Time Course Network Enrichment
We developed time course network enrichment (TiCoNE), a novel human-augmented time series clustering method combined with a temporal network enricher that enables drug target discovery based on temporal networks. Temporal gene clusters are embedded into molecular networks, and TiCoNE identifies molecular pathways (subnetworks) with a differential behavior under two conditions (e.g., diseases). Such temporal disease pathway candidates are evaluated by calculating empirical p-values.
TiCoNE works with most kinds of biological entities (genes, proteins, RNAs, etc.) and most types of molecular measures acquirable for them (transcriptomics,proteomics, etc.). It comes as web server and as Cytoscape plugin.
Web: https://ticone.compbio.sdu.dk/
Papers:
-
C. Wiwie et al., “Time-resolved systems medicine reveals viral infection-modulating host targets,” Syst. Med., vol. 2, no. 1, 2019.
ClustEval - The Integrative Clustering Evaluation Framework
ClustEval is a free and extendable opensource platform for objective performance comparison of arbitrary Clustering Methods on different datasets. It is designed to support the standard processes related to Cluster Analyses.
Thanks to highly advanced wet-lab techniques, we are produceing a tremndous amount of biological data everyday. This wealth of data urges for sophisticated automatic knowledge extraction techniques. One of the most popular techniques is clustering, i.e., the grouping of similar objects into clusters.
Even though clustering is a long standing problem in computer science, conducting a high-quality cluster analysis is all but straight forward. For the practitioner, the very plethora of existing clustering algorithms is already a huge obstacle. Each tool requires at least one parameter and does not perform equally well on every dataset. Finding the optimal tool and paramter setting is a very tiresome and error-prone process.
With ClustEval, we introduce an integrated clustering framework, assisting the user in all steps of cluster analyses, from data preprocessing and parameter optimization to evaluating the reported clusters. The framework allows the fully automated execution of many different tool on given datasets and an exhaustive evaluation. The flexibility of the framework allows convenient extension with new tools, datasets, and quality measures. Furthermore, the website summarizes the results of millions of cluster evaluations providing an excellent overview of the current state-of-the-art clustering tools.
Web: http://clusteval.sdu.dk
Papers:
-
Wiwie C et al. Comparing the performance of biomedical clustering methods. Nature Methods 2015, doi:10.1038/nmeth.3583.
Going large-scale - Transitivity Clustering with missing values

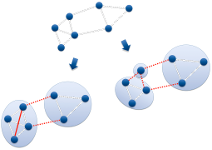
Next generation sequencing as well as other high-throughput OMICS technologies are constantly feeding a trend in data growth of available biological data. To date, the National Center for Biotechnology Information (NCBI) lists over 2,000 completed and annotated genome sequences together with several thousands of ongoing projects. However, the knowledge of the organism's sequence alone is by far not enough to fully understand an organism in all facets of its behavior. One widely used computational approach for unraveling knowledge is evolutionary homology detection, typically carried out by means of clustering a given similarity matrix. Calculating the necessary similarities between all pairs of objects (proteins in this case) poses the current bottleneck of all clustering approaches. In this project, we developed a data partitioning tool that builts on Transitivity Clustering, which is capable of dealing with missing values but still robust in clustering quality.
Web: http://transclust.mpi-inf.mpg.de
Papers:
-
Roettger R et al. Online Transitivity Clustering of Biological Data with Missing Values. Proceedings of the German Conference on Bioinformatics 2012, OpenAccess Series in Informatics, 26:57-68.
-
Wittkop T et al. Partitioning biological data with Transitivity Clustering. Nature Methods 2010 Jun;7(6):419-20.
-
Wittkop T et al. Extension and robustness of Transitivity Clustering for protein-protein interaction network analysis. Int Math. 2011, 7:4, 255-273.
Drug target discovery & vaccine design - The evolution of actinobacteria
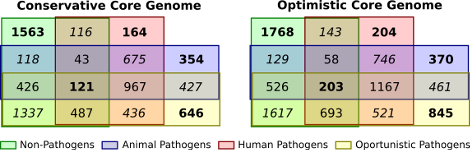
For identifying potential drug targets, information about evolutionarily conserved proteins or protein families is very helpful. It is desirable to identify exo-proteins that are only conserved amongst pathogenic species but not in non-pathogens. These are potential drug targets because attacking these proteins would leave useful bacteria largely untouched. In order to detect such proteins, we applied Transitivity Clustering for remote homology detection of proteins of 89 different actinobacteria, among these the pathogenic agents for tuberculosis and diptheria, for instance. A challenge for all clustering tools is the detection of a meaningful density parameter, especially if no gold standard data is available. Here, we developed a measure for judging the quality of a clustering result based by exploiting only intrinsic properties of the given datasets.
In this project we collaborate with Vasco Azevedo from the Federal University of Minas Gerais, Belo Horizonte, Brazil.
Web: http://transclust.mpi-inf.mpg.de
Papers:
- Roettger R et al. Density Parameter Estimation for Finding Clusters of Homologous Proteins - Tracing Actinobacterial Pathogenicity Life Styles. Bioinformatics Bioinformatics. 2013 Jan 15;29(2):215-22.
- Roettger R et al. Online Transitivity Clustering of Biological Data with Missing Values. Proceedings of the German Conference on Bioinformatics 2012, OpenAccess Series in Informatics, 26:57-68.
Drug target prediction - Integrative models of gene regulatory networks

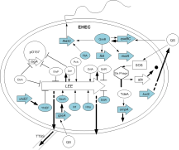
The increasing in genome sequencing will provide us with thousands of sequenced microbial organisms over the next years. However, this is only the first step in understanding how cells survive, reproduce and adapt their behavior while being exposed to changing environmental conditions. One major control mechanism is transcriptional gene regulation. Here, striking is the direct juxtaposition of the handful of bacterial model organisms to the >2,000 prokaryotic genomes that we have available for download from NCBI. The main idea is to use the known transcriptional regulatory network of reference organisms as template in order to unravel evolutionarily conserved gene regulations in newly sequenced species.
Web: http://www.coryneregnet.de, http://www.ehecregnet.de
Papers:
- Pauling J et al. On the trail of EHEC/EAEC - Unraveling the gene regulatory networks of human pathogenic Escherichia coli bacteria. Integr Biol., 2012, 4 (7), 728 - 733.
- Pauling J et al. CoryneRegNet 6.0 - Updated database content, new analysis methods and novel features focusing on community demands. Nucleic Acids Res. 2012 Jan;40(1):D610-4.